

CLASSIFICATION OF MITOCHONDRIAL DISEASES
The classification of mitochondrial diseases reflects
the peculiar feature of the oxidative phosphorylation system of being formed by
proteins encoded by two different genomes: mtDNA and nuclear genome.
The first group of illnesses is characterized by the presence of mutations in mtDNA, that can be transmitted either maternally or sporadic. A second group of disorders is caused by mutations in nuclear genes that make up or control oxidative phosphorylation (OXPHOS). Since many OXPHOS-related nuclear genes are still unknown in humans, several of these illnesses are classified only on the basis of biochemical alterations, revealed by the analysis of affected tissue (especially skeletal muscle). The high clinical, biochemical and molecular heterogeneity of these disorders explains why only about 50% of the patients with a clinical/biochemical defined mitochondrial disease have a genetic diagnosis.
The recent technological advances in the field of next-generation DNA sequencing (NGS) offer an affordable and highly informative tool for the rapid and cost-effective analysis of a large number of DNA sequences at the same time. This approach has led to the identification of several nuclear genes associated with mitochondrial diseases and to an improvement in the diagnostic process.
1. MtDNA Mutations
This group includes syndromes caused by either mtDNA point mutations, large-scale rearrangements of mtDNA or reduction in the mtDNA amount.
1.1 Point mutations
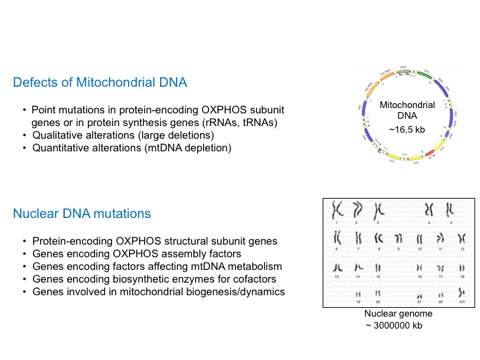
These are clinical entities associated with the substitution of single bases or micro-insertions/micro-deletions in the mtDNA molecule. These mutations may concern genes encoding transfer RNAs (tRNA), ribosomal RNAs, (rRNA), or messenger RNAs (mRNA) that are then translated into proteins. Almost all mtDNA point mutations are maternally transmitted. They are often, but not always, heteroplasmic. Even if more than 100 point mutations have been described in association with an extremely heterogeneous spectrum of clinical presentations, only a few of them are frequent and associated with well-defined clinical syndromes.
Leber's Hereditary Optic Neuropathy (LHON, OMIM535000) is a juvenile-onset condition affecting mostly males. It is characterized by acute or sub-acute loss of central vision due to rapidly progressive optic atrophy. This partial or complete (and usually permanent) loss of vision is the only consistent manifestation of the disease which, more rarely, may also include alterations in cardiac rhythm (ventricular pre-excitation syndrome). The muscle biopsy does not show evidence of ragged-red fibers and is not necessary for the diagnosis of the disease. This disease is associated with mutations in the nucleotide positions m.3460, 11778, or 14484 of mtDNA, in the gene encoding subunits ND1, ND4, and ND6 of complex I, respectively. Other mutations, all present in complex I mtDNA genes, have recently been identified. Many features of LHON remain to be clarified, including the extreme tissue specificity of the anatomical and clinical lesion, the prevalence among males, and the biochemical consequences of each mutation.
Neurogenic muscle weakness, ataxia, retinitis pigmentosa (NARP, OMIM551500) can also include, besides the above-mentioned symptoms, epilepsy, and sometimes mental deterioration. Symptoms usually appear in adulthood. Ragged-red fibers are absent in the muscle biopsy. The disease is associated with mutation m. 8993T>G in the gene encoding subunit 6 of mitochondrial ATPase (complex V of the respiratory chain). In patients presenting a less serious NARP phenotype, a transition T->C in the same position has also been described. The same m. 8993T>G mutation when present in >90% of total mitochondrial genomes, leads to the more severe, earlier onset Leigh syndrome (MILS, maternally inherited Leigh syndrome).
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes, (MELAS, OMIM540000) is defined by the following symptoms: 1) stroke-like episodes caused by focal cerebral lesions, often localized in the parieto-occipital regions of the brain; 2) lactic acidosis or abnormal lactic levels in the blood (and cerebro-spinal fluid, CSF); 3) "ragged-red" fibers in the muscle biopsy. Other signs involving the central nervous system include mental deterioration, recurrent migraine with "cerebral" vomiting, focal or generalized epilepsy and neurosensory deafness. The disease is transmitted maternally and the onset varies from early childhood to young adulthood. MELAS syndrome is typically associated with mutation m.3243A>G in the gene encoding tRNA-Leu(UUR). Other point mutations associated with MELAS have been reported, although they are much rarer than the m.3243A>G.
Myoclonus Epilepsy with Ragged-Red Fibers, (MERRF, OMIM545000) is characterized by the association of myoclonus, epilepsy, muscle weakness and wasting, motor incoordination (ataxia) and sometimes, mental deterioration. Clinical manifestations can vary greatly even within the same family. This variation is attributed to the quantity of mutated mtDNA in relation to normal mtDNA (heteroplasmy) and to variation in the tissue distribution of the mutation. The major part of affected families carry an m.8344A>G transition in the gene encoding tRNA-Lys.
Numerous other point mutations of mtDNA have been associated with different clinical phenotypes in single patients or in a few families.
1.2 Qualitative alterations of mtDNA
These can be either partial deletions of mtDNA or less frequently, partial duplications. Both types are heteroplasmic since they co-exist with normal mtDNA. These coarse mtDNA alterations are almost invariably associated with 3 main clinical presentations: Kearns-Sayre Syndrome, Progressive External Ophthalmoplegia and Pearson's Syndrome. Kearns-Sayre Syndrome is a serious illness that occurs sporadically, and includes the triad of (1) Progressive External Ophthalmoplegia (PEO) with bilateral drooping eyelids (ptosis), (2) Pigmentary Retinopathy, and (3) onset before twenty years of age. Other frequent signs are poor growth, motor incoordination (ataxia) due to cerebellar failure, mental deterioration, deafness, and alterations of cardiac rhythm (Atrio-ventricular blocks) often requiring the application of a pace-maker.
Pearson's Syndrome is a rare sporadic disease affecting newborn or very young babies. It is characterized by sideroblastic anemia, pancytopenia and failure of exocrine pancreas with intestinal malabsorption. A progressive improvement in the hematologic and gastro-intestinal situation takes place in children who survive the first years, but they usually develop a typical Kearns-Sayre Syndrome afterwards.
Progressive External Ophthalmoplegia (PEO) also occurs sporadically and is characterized by the appearance of bi-lateral ptosis and paralysis of eye-movement, often associated with weakness in girdle muscles of the shoulders and pelvis. It appears in adulthood. PEO is often associated with mtDNA multiple deletions. Mutations in diverse nuclear genes have been associated with autosomal dominant or recessive PEO (see above Mitochondriopathy due to mutations in nuclear genes).
1.3 Quantitative alterations of mtDNA
A reduction of the amount of mtDNA is called "depletion". Usually these forms are infantile with a progressive course. The most involved organs are: skeletal muscle and heart, liver and brain. Depletions of the mtDNA are caused by mutations in the nuclear genes (see above "Mitochondriopathy due to mutations in nuclear genes").
2. Mitochondriopathy due to mutations in nuclear genes
Over 90% of mitochondrial proteins are expressions of nuclear genes and include the majority of the components of the OXPHOS system, proteins necessary for the assembly of the respiratory chain complexes. Moreover mtDNA replication, transcription and translation are dependent on nuclear encoded-proteins. A growing number of degenerative hereditary diseases, especially in the neurological field, have been linked to mutations in genes encoding proteins that enter the mitochondrion and are more or less directly correlated to OXPHOS.
The following is a brief outline of only one disease, Leigh's Syndrome, the most common and well-known of the group of diseases that result from abnormalities in nuclear genes related to mitochondrial OXPHOS (although, as reported in the paragraph 1.1 in about 20% of cases there are mutations in the ATPase 6 gene and occasionally also point mutations in mtDNA genes encoding COX subunits or tRNAs). After an initial period of normal development in the first months of infancy, affected children present a progressive delay in psychomotor development, accompanied by incoordination of eye movements, recurrent vomiting, epilepsy, abnormalities in breathing rate, and lactic acidosis. These symptoms can be referred to symmetric lesions of neurological structures that originate, cross through, or are localized in the basal ganglia, brainstem and cerebellum.
The frequent increase in lactic acid levels in blood and CSF suggests an alteration in mitochondria energy metabolism. In more than half the cases it is possible to document a genetic alteration. In about 30% of the cases the biochemical defect is a profound decrease in the activity of Complex IV activity (cytochrome c oxidase, COX) and the genetic alteration is due, in most cases, to mutations in an assembly gene of complex IV, called SURF1. In other cases, the biochemical defect is found in Complex I or Complex II of the respiratory chain. Mutations in the subunits of these complexes have been identified in some patients. Finally, in 10% of the cases a deficiency in Pyruvate Dehydrogenase is detected, usually associated with mutations in the X-linked gene encoding subunit E1-alpha of the enzyme.
A grouping of the corresponding proteins based on their biological role among:
can be used for the classification of mitochondrial disorders due to mutations in nuclear genes.
There are some proteins, not included in this classification but indirectly correlated to OXPHOS, whose mutations cause mitochondriopathies: e.g. proteins necessary for protein import into mitochondria (TIMM8A, DNAJC19), proteins linked to apoptosis (AIFM1), and proteins with a detoxifying role (ETHE1). All these conditions are extremely rare.
2.1 Disorders due to defects in nuclear gene encoding structural components of the OXPHOS complexes
Although 72 of the 85 subunits of the OXPHOS system are encoded by nuclear DNA, mutations of these genes have only rarely been described. This could imply that such mutations are highly deleterious and probably embryo-lethal. Mutations that have been described in fact are usually associated to a neonatal or early-onset, although occasional patients with a late onset of disease have been reported. On the other hand the screening of the nuclear-encoded subunits of respiratory chain has not always been done in a systematic manner, especially for complex I. Thanks to the introduction of NGS technologies, the number of reports regarding mutations in structural OXPHOS component is grown up. The mutations in nuclear encoded subunits identified so far, are mainly abnormalities of complex I found in patients with- infancy or childhood-onset, even if also mutations in structural subunits of complex II, III, IV and V were described. Typical phenotypic presentations are: Leigh syndrome, leukoencephalopathy and cardio-myopathy.
2.2 Respiratory chain complex assembly deficiencies
OXPHOS complexes are multiheteromeric structure and for their proper assembly several different factors are required; mutations affecting these factors lead to formation of instable or partly functioning complexes. For complexes III, IV and V, genetic alterations in genes belonging to this group are more frequent that the mutations affecting structural subunits, reported in the previous paragraph. The main clinical presentations are the same, with various kinds of encephalomyopathies.
2.3 Disorders due to gene defects altering the mtDNA maintenance
MtDNA remains dependent upon nuclear genome for the production of proteins involved in its replication, transcription, translation, repair and maintenance. Replication of mtDNA requires a small set of proteins: i.e. the DNA polymerase gamma (POLG), Twinkle helicase (PEO1), mitochondrial single-stranded DNA binding protein (mtSSB), and a supply of deoxy-nucleotides triphosphate (dNTPs). Structural defects of the DNA-processive enzymes are often associated with mtDNA mutagenesis and multiple mtDNA deletions (qualitative alterations; see paragraph 1.2), whereas defects affecting the dNTP pool usually cause mtDNA depletion, that mean reduction of mtDNA copy number/cell (quantitative alterations; see paragraph 1.3).
Progressive External Ophthalmoplegia and muscle weakness are typical of genes causing mtDNA qualitative alterations while genes associated with quantitative alterations (mtDNA depletions) cause different disorders with neurological, muscular or hepatic involvement.
Mitochondrial DNA translation or protein synthesis is carried out in the mitochondrial matrix by a machinery, which is composed of tRNAs and rRNAs synthesized in situ from the corresponding mitochondrial genes and a number of proteins encoded by nuclear DNA and imported into mitochondria. Different mutations affecting subunits of mitochondrial ribosomes, enzymes with a role in mitochondrial tRNA maturation, elongation factors, amino acyl tRNA synthetases have been found in patients with mitochondrial diseases and clinically constitute one of the most heterogeneous groups.
2.4 Defects of genes encoding factors involved in the biosynthesis of cofactors
CoQ or ubiquinone is a lipophilic component of the electron-transport chain, which transfers electrons from Complex I or II, and from the oxidation of fatty acids and branched-chain amino acids, to Complex III. The CoQ also plays a role as an antioxidant and as a membrane stabilizer. Mutations in genes encoding enzymes responsible for the CoQ biosynthesis are usually associated with CoQ10 deficiency in muscle and cause myoglobinuria and/or early-onset ataxia. All components of the respiratory chain are embedded in the lipid milieu of the inner mitochondrial membrane, which is composed predominantly of cardiolipin. Cardiolipin is not merely a scaffold but is essential for proper functioning of several mitochondrial OXPHOS complexes and several mitochondrial carrier proteins. This is the reason why defects in cardiolipin could cause OXPHOS dysfunction and hence mitochondrial disease. In fact there is an example on this regard, the Barth syndrome (mitochondrial myopathy, cardiomyopathy, growth retardation, and leukopenia), caused by mutation in the gene TAZ o tafazzin.
The OXPHOS complexes need to be equipped with cofactors such as copper, heme or iron-sulphur (FeS) clusters that are necessary for their electron transport capacity. A whole range of complex-specific chaperones, assembly factors and enzymes involved in the biosynthesis and incorporation of prosthetic groups are necessary for the assembly of intact and enzymatically functional complexes. The clinical spectrum associated with these impairments is broad: myopathy, leukoencephalopathy, sideroblastic anemia
2.5 Defects of proteins involved in mitochondrial biogenesis/dynamics
Mitochondria are not static and isolated organelles but form a complex network. Mitochondrial fusion and fission require conserved protein machineries at the outer and inner membranes that mediate membrane mixing and division events. The primary protein for the regulation of inner membrane morphology is OPA1, mutations of which cause optic atrophy. Neuropathies, Charcot-Marie-Tooth type, are instead associated with mutations in MFN2, fusion protein of the outer membrane, while mutation in genes encoding mitochondrial fission factors (DNM1L, MFF) are usually responsible for severe, early-onset encephalopathies.
The first group of illnesses is characterized by the presence of mutations in mtDNA, that can be transmitted either maternally or sporadic. A second group of disorders is caused by mutations in nuclear genes that make up or control oxidative phosphorylation (OXPHOS). Since many OXPHOS-related nuclear genes are still unknown in humans, several of these illnesses are classified only on the basis of biochemical alterations, revealed by the analysis of affected tissue (especially skeletal muscle). The high clinical, biochemical and molecular heterogeneity of these disorders explains why only about 50% of the patients with a clinical/biochemical defined mitochondrial disease have a genetic diagnosis.
The recent technological advances in the field of next-generation DNA sequencing (NGS) offer an affordable and highly informative tool for the rapid and cost-effective analysis of a large number of DNA sequences at the same time. This approach has led to the identification of several nuclear genes associated with mitochondrial diseases and to an improvement in the diagnostic process.
1. MtDNA Mutations
This group includes syndromes caused by either mtDNA point mutations, large-scale rearrangements of mtDNA or reduction in the mtDNA amount.
1.1 Point mutations
These are clinical entities associated with the substitution of single bases or micro-insertions/micro-deletions in the mtDNA molecule. These mutations may concern genes encoding transfer RNAs (tRNA), ribosomal RNAs, (rRNA), or messenger RNAs (mRNA) that are then translated into proteins. Almost all mtDNA point mutations are maternally transmitted. They are often, but not always, heteroplasmic. Even if more than 100 point mutations have been described in association with an extremely heterogeneous spectrum of clinical presentations, only a few of them are frequent and associated with well-defined clinical syndromes.
Leber's Hereditary Optic Neuropathy (LHON, OMIM535000) is a juvenile-onset condition affecting mostly males. It is characterized by acute or sub-acute loss of central vision due to rapidly progressive optic atrophy. This partial or complete (and usually permanent) loss of vision is the only consistent manifestation of the disease which, more rarely, may also include alterations in cardiac rhythm (ventricular pre-excitation syndrome). The muscle biopsy does not show evidence of ragged-red fibers and is not necessary for the diagnosis of the disease. This disease is associated with mutations in the nucleotide positions m.3460, 11778, or 14484 of mtDNA, in the gene encoding subunits ND1, ND4, and ND6 of complex I, respectively. Other mutations, all present in complex I mtDNA genes, have recently been identified. Many features of LHON remain to be clarified, including the extreme tissue specificity of the anatomical and clinical lesion, the prevalence among males, and the biochemical consequences of each mutation.
Neurogenic muscle weakness, ataxia, retinitis pigmentosa (NARP, OMIM551500) can also include, besides the above-mentioned symptoms, epilepsy, and sometimes mental deterioration. Symptoms usually appear in adulthood. Ragged-red fibers are absent in the muscle biopsy. The disease is associated with mutation m. 8993T>G in the gene encoding subunit 6 of mitochondrial ATPase (complex V of the respiratory chain). In patients presenting a less serious NARP phenotype, a transition T->C in the same position has also been described. The same m. 8993T>G mutation when present in >90% of total mitochondrial genomes, leads to the more severe, earlier onset Leigh syndrome (MILS, maternally inherited Leigh syndrome).
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes, (MELAS, OMIM540000) is defined by the following symptoms: 1) stroke-like episodes caused by focal cerebral lesions, often localized in the parieto-occipital regions of the brain; 2) lactic acidosis or abnormal lactic levels in the blood (and cerebro-spinal fluid, CSF); 3) "ragged-red" fibers in the muscle biopsy. Other signs involving the central nervous system include mental deterioration, recurrent migraine with "cerebral" vomiting, focal or generalized epilepsy and neurosensory deafness. The disease is transmitted maternally and the onset varies from early childhood to young adulthood. MELAS syndrome is typically associated with mutation m.3243A>G in the gene encoding tRNA-Leu(UUR). Other point mutations associated with MELAS have been reported, although they are much rarer than the m.3243A>G.
Myoclonus Epilepsy with Ragged-Red Fibers, (MERRF, OMIM545000) is characterized by the association of myoclonus, epilepsy, muscle weakness and wasting, motor incoordination (ataxia) and sometimes, mental deterioration. Clinical manifestations can vary greatly even within the same family. This variation is attributed to the quantity of mutated mtDNA in relation to normal mtDNA (heteroplasmy) and to variation in the tissue distribution of the mutation. The major part of affected families carry an m.8344A>G transition in the gene encoding tRNA-Lys.
Numerous other point mutations of mtDNA have been associated with different clinical phenotypes in single patients or in a few families.
1.2 Qualitative alterations of mtDNA
These can be either partial deletions of mtDNA or less frequently, partial duplications. Both types are heteroplasmic since they co-exist with normal mtDNA. These coarse mtDNA alterations are almost invariably associated with 3 main clinical presentations: Kearns-Sayre Syndrome, Progressive External Ophthalmoplegia and Pearson's Syndrome. Kearns-Sayre Syndrome is a serious illness that occurs sporadically, and includes the triad of (1) Progressive External Ophthalmoplegia (PEO) with bilateral drooping eyelids (ptosis), (2) Pigmentary Retinopathy, and (3) onset before twenty years of age. Other frequent signs are poor growth, motor incoordination (ataxia) due to cerebellar failure, mental deterioration, deafness, and alterations of cardiac rhythm (Atrio-ventricular blocks) often requiring the application of a pace-maker.
Pearson's Syndrome is a rare sporadic disease affecting newborn or very young babies. It is characterized by sideroblastic anemia, pancytopenia and failure of exocrine pancreas with intestinal malabsorption. A progressive improvement in the hematologic and gastro-intestinal situation takes place in children who survive the first years, but they usually develop a typical Kearns-Sayre Syndrome afterwards.
Progressive External Ophthalmoplegia (PEO) also occurs sporadically and is characterized by the appearance of bi-lateral ptosis and paralysis of eye-movement, often associated with weakness in girdle muscles of the shoulders and pelvis. It appears in adulthood. PEO is often associated with mtDNA multiple deletions. Mutations in diverse nuclear genes have been associated with autosomal dominant or recessive PEO (see above Mitochondriopathy due to mutations in nuclear genes).
1.3 Quantitative alterations of mtDNA
A reduction of the amount of mtDNA is called "depletion". Usually these forms are infantile with a progressive course. The most involved organs are: skeletal muscle and heart, liver and brain. Depletions of the mtDNA are caused by mutations in the nuclear genes (see above "Mitochondriopathy due to mutations in nuclear genes").
2. Mitochondriopathy due to mutations in nuclear genes
Over 90% of mitochondrial proteins are expressions of nuclear genes and include the majority of the components of the OXPHOS system, proteins necessary for the assembly of the respiratory chain complexes. Moreover mtDNA replication, transcription and translation are dependent on nuclear encoded-proteins. A growing number of degenerative hereditary diseases, especially in the neurological field, have been linked to mutations in genes encoding proteins that enter the mitochondrion and are more or less directly correlated to OXPHOS.
The following is a brief outline of only one disease, Leigh's Syndrome, the most common and well-known of the group of diseases that result from abnormalities in nuclear genes related to mitochondrial OXPHOS (although, as reported in the paragraph 1.1 in about 20% of cases there are mutations in the ATPase 6 gene and occasionally also point mutations in mtDNA genes encoding COX subunits or tRNAs). After an initial period of normal development in the first months of infancy, affected children present a progressive delay in psychomotor development, accompanied by incoordination of eye movements, recurrent vomiting, epilepsy, abnormalities in breathing rate, and lactic acidosis. These symptoms can be referred to symmetric lesions of neurological structures that originate, cross through, or are localized in the basal ganglia, brainstem and cerebellum.
The frequent increase in lactic acid levels in blood and CSF suggests an alteration in mitochondria energy metabolism. In more than half the cases it is possible to document a genetic alteration. In about 30% of the cases the biochemical defect is a profound decrease in the activity of Complex IV activity (cytochrome c oxidase, COX) and the genetic alteration is due, in most cases, to mutations in an assembly gene of complex IV, called SURF1. In other cases, the biochemical defect is found in Complex I or Complex II of the respiratory chain. Mutations in the subunits of these complexes have been identified in some patients. Finally, in 10% of the cases a deficiency in Pyruvate Dehydrogenase is detected, usually associated with mutations in the X-linked gene encoding subunit E1-alpha of the enzyme.
A grouping of the corresponding proteins based on their biological role among:
can be used for the classification of mitochondrial disorders due to mutations in nuclear genes.
There are some proteins, not included in this classification but indirectly correlated to OXPHOS, whose mutations cause mitochondriopathies: e.g. proteins necessary for protein import into mitochondria (TIMM8A, DNAJC19), proteins linked to apoptosis (AIFM1), and proteins with a detoxifying role (ETHE1). All these conditions are extremely rare.
2.1 Disorders due to defects in nuclear gene encoding structural components of the OXPHOS complexes
Although 72 of the 85 subunits of the OXPHOS system are encoded by nuclear DNA, mutations of these genes have only rarely been described. This could imply that such mutations are highly deleterious and probably embryo-lethal. Mutations that have been described in fact are usually associated to a neonatal or early-onset, although occasional patients with a late onset of disease have been reported. On the other hand the screening of the nuclear-encoded subunits of respiratory chain has not always been done in a systematic manner, especially for complex I. Thanks to the introduction of NGS technologies, the number of reports regarding mutations in structural OXPHOS component is grown up. The mutations in nuclear encoded subunits identified so far, are mainly abnormalities of complex I found in patients with- infancy or childhood-onset, even if also mutations in structural subunits of complex II, III, IV and V were described. Typical phenotypic presentations are: Leigh syndrome, leukoencephalopathy and cardio-myopathy.
2.2 Respiratory chain complex assembly deficiencies
OXPHOS complexes are multiheteromeric structure and for their proper assembly several different factors are required; mutations affecting these factors lead to formation of instable or partly functioning complexes. For complexes III, IV and V, genetic alterations in genes belonging to this group are more frequent that the mutations affecting structural subunits, reported in the previous paragraph. The main clinical presentations are the same, with various kinds of encephalomyopathies.
2.3 Disorders due to gene defects altering the mtDNA maintenance
MtDNA remains dependent upon nuclear genome for the production of proteins involved in its replication, transcription, translation, repair and maintenance. Replication of mtDNA requires a small set of proteins: i.e. the DNA polymerase gamma (POLG), Twinkle helicase (PEO1), mitochondrial single-stranded DNA binding protein (mtSSB), and a supply of deoxy-nucleotides triphosphate (dNTPs). Structural defects of the DNA-processive enzymes are often associated with mtDNA mutagenesis and multiple mtDNA deletions (qualitative alterations; see paragraph 1.2), whereas defects affecting the dNTP pool usually cause mtDNA depletion, that mean reduction of mtDNA copy number/cell (quantitative alterations; see paragraph 1.3).
Progressive External Ophthalmoplegia and muscle weakness are typical of genes causing mtDNA qualitative alterations while genes associated with quantitative alterations (mtDNA depletions) cause different disorders with neurological, muscular or hepatic involvement.
Mitochondrial DNA translation or protein synthesis is carried out in the mitochondrial matrix by a machinery, which is composed of tRNAs and rRNAs synthesized in situ from the corresponding mitochondrial genes and a number of proteins encoded by nuclear DNA and imported into mitochondria. Different mutations affecting subunits of mitochondrial ribosomes, enzymes with a role in mitochondrial tRNA maturation, elongation factors, amino acyl tRNA synthetases have been found in patients with mitochondrial diseases and clinically constitute one of the most heterogeneous groups.
2.4 Defects of genes encoding factors involved in the biosynthesis of cofactors
CoQ or ubiquinone is a lipophilic component of the electron-transport chain, which transfers electrons from Complex I or II, and from the oxidation of fatty acids and branched-chain amino acids, to Complex III. The CoQ also plays a role as an antioxidant and as a membrane stabilizer. Mutations in genes encoding enzymes responsible for the CoQ biosynthesis are usually associated with CoQ10 deficiency in muscle and cause myoglobinuria and/or early-onset ataxia. All components of the respiratory chain are embedded in the lipid milieu of the inner mitochondrial membrane, which is composed predominantly of cardiolipin. Cardiolipin is not merely a scaffold but is essential for proper functioning of several mitochondrial OXPHOS complexes and several mitochondrial carrier proteins. This is the reason why defects in cardiolipin could cause OXPHOS dysfunction and hence mitochondrial disease. In fact there is an example on this regard, the Barth syndrome (mitochondrial myopathy, cardiomyopathy, growth retardation, and leukopenia), caused by mutation in the gene TAZ o tafazzin.
The OXPHOS complexes need to be equipped with cofactors such as copper, heme or iron-sulphur (FeS) clusters that are necessary for their electron transport capacity. A whole range of complex-specific chaperones, assembly factors and enzymes involved in the biosynthesis and incorporation of prosthetic groups are necessary for the assembly of intact and enzymatically functional complexes. The clinical spectrum associated with these impairments is broad: myopathy, leukoencephalopathy, sideroblastic anemia
2.5 Defects of proteins involved in mitochondrial biogenesis/dynamics
Mitochondria are not static and isolated organelles but form a complex network. Mitochondrial fusion and fission require conserved protein machineries at the outer and inner membranes that mediate membrane mixing and division events. The primary protein for the regulation of inner membrane morphology is OPA1, mutations of which cause optic atrophy. Neuropathies, Charcot-Marie-Tooth type, are instead associated with mutations in MFN2, fusion protein of the outer membrane, while mutation in genes encoding mitochondrial fission factors (DNM1L, MFF) are usually responsible for severe, early-onset encephalopathies.

Mutation map of mtDNA.